Historical Evaluation of Pharmacovigilance
Historical Evaluation of Pharmacovigilance
Historical Evaluation of Pharmacovigilance: We would be going through the origin and evolution of Pharmacovigilance to its present stage.
Let’s start!

On the 29th of January 1848, Hannah Greener, from Winlaton, England, underwent a routine procedure under general anesthesia using chloroform for the treatment of an ingrowing toenail.
She was a healthy 15-yr-old girl who underwent the removal of another toenail successfully using the anesthetic agent diethyl ether several months prior to this.
Chloroform, an anesthetic agent, was introduced into clinical practice a year earlier by James Simpson, professor of midwifery at Edinburgh.
Hannah used chloroform since it produced less nausea and vomiting than ether.
Following other deaths and continuing concerns of the public and clinicians about the safety of chloroform as an anesthetic, the Lancet Journal created a commission to tackle this problem.
At that point in time, the safety of chloroform as a general anesthetic became controversial all over the world.
1889: THE LANCET appointed Dr. Lauder Brunton (1844-1916) as a consultant on the second Hyderabad Chloroform commission.
This commission invited doctors in Britain and its colonies to report anesthesia-related deaths.
1893: results subsequently published in The Lancet
Thus resulting in the first trigger for the reporting of Spontaneous reports for suspected adverse drug reactions (ADR).
Since that episode, advances in drug regulation often seem to be provoked by misfortune, or even disaster.
1905: Samuel Hopkins Adams (America) published “The Great American Fraud,” in which he exposed many of the false claims made about patent medicines.
30 June 1906: The US Federal Food and Drug Act was formulated and it required that the drugs be clean and free of contamination, but there was nothing mentioned regarding the effectiveness.
Furthermore, in 1911, this organization banned false therapeutic indications of drugs.
1937 (United States): a liquid form of sulfanilamide (a sulfonamide indicated for streptococcal infections) came into the drug market for pediatric patients.
- The drug was distributed in tablet and powder form without serious reactions
- Sulfanilamide liquid formulation reportedly resulted in more than 100 deaths in the United States
- Sulfanilamide dissolved in diethyl glycol which was a highly toxic chemical analogue of antifreeze
Consequently, the Federal Food, Drug, and Cosmetic Act was formulated on 25 June 1938 to modernize the public health system.
This law brought cosmetics and medical devices under control, and it required that the drugs be labeled with adequate directions for safe use.
Moreover, it mandated pre-market approval of all new drugs, such that a manufacturer would have to prove to the FDA that a drug was safe before it could be sold.
Again in 1938, a physician (Douthwaite) described that acetylsalicylic acid (ASA) could cause melaena.
1952: Cases of hematotoxicity (aplastic anemia) associated with the use of chloramphenicol were reported in the United States.
As a result, the Council on Drugs of the American Medical Association set up a Blood Dyscrasia Registry on 23 October 1952.

The next big disaster has happened with Thalidomide.
- Thalidomide first synthesized in 1954.
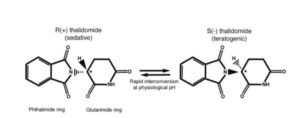
THALIDOMIDE – Developmental toxicology - Thalidomide first marketed in Western Germany as a sedative and hypnotic agent in 1956 and sold as an over-the-counter remedy.
- Widely prescribed as a harmless treatment for morning sickness in pregnant women.
- The pharmaceutical company advertised their product as “completely safe” for everyone, including mother and child, “even during pregnancy,” as the manufacturer could not find a dose high enough to kill a rat.
- By 1960, thalidomide marketed and distributed in 46 countries around the world using different brand names.
- At the same time, Australian obstetrician Dr. William McBride observed that the drug also alleviated morning sickness and prescribed for off-label use.
- Peripheral neuropathy and severe birth defects were reported by the patients after taking the drug.

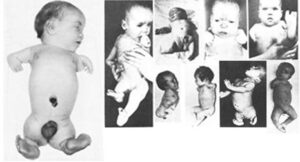
1961: Two independent clinicians, Lenz in Germany and McBride in Australia, confirmed that thalidomide causes severe birth defects in children in over 10,000 cases.
Many children were born with phocomelia as a side effect of the drug thalidomide which resulted in the shortening or absence of limbs.
There was also an increase in the reports of miscarriage rates during this same period, thus resulting in man‐made medical disaster.
By 25th November 1961, thalidomide was withdrawn from the market by its manufacturer.

After the Thalidomide disaster
By 1961, the Food and Drug Administration (FDA) started the systematic collection of reports of all types of adverse drug reactions, mainly through the Hospital Reporting Program.
1962 (USA): the Drug Amendments Act was passed by Congress which required the submission of safety and efficacy data of drugs prior to marketing so as to approve all new drug applications.
As a result of this amendment, the safety data required to additionally include teratogenicity tests in three different animals.
1963: As a result of the thalidomide tragedy, the whole regulatory system reformed in the United Kingdom (UK) where a Committee on the Safety of Drugs (CSD) started.
Following this, a voluntary adverse drug reaction reporting system (Yellow Card Scheme) came into being in the UK (1964).
In Europe (1965), It stimulated the development of European legislation with the EC Directive 65/65 following the tragedy of Thalidomide, which was the first of its kind and ensuring a high level of protection for public health.
1966: Boston Collaborative Drug Surveillance Program started. The first group to conduct epidemiological research to quantify the potential adverse effects of drugs.
1968: The WHO’s started the Programme for International Drug Monitoring as a Pilot Research Project.
Ten member countries participated in this program to pool the adverse drug reactions.
1973: A retrospective study showed the correlation between the congenital malformations of babies and the ingestion of thalidomide during pregnancy.
Council for International Organizations of Medical Sciences (CIOMS) functioned as a forum for discussion between international drug regulatory authorities and pharmaceutical companies since 1977.
1978: Establishment of the Uppsala Monitoring Centre (UMC) to support the WHO Programme for International Drug Monitoring.
1986: CIOMS set up a working group on International Reporting of Adverse Drug Reactions.
1990: Establishment of the International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH).
(A collaborative initiative between the EU, Japan, and the United States with observers from WHO, European Free Trade Association (EFTA), and Canada)
1992: The European Society of Pharmacovigilance (ESoP) turned into the International Society of Pharmacovigilance (IsoP).
1993 – Establishment of MedWatch to collect the data regarding adverse events.
October 1994 – ICH adopted MedDRA Version 1.0 as a basis for international terminology.
1995: Establishment of the European Medicines Agency (EMA).
17 July 1997 – Endorsed the first ICH E2B guideline i.e. Data Elements for Transmission of Individual Case Safety Reports.
1998: UMC implemented an automated signal detection method, using a Bayesian Confidence Propagation Neural Network (BCPNN) data mining approach.
2001: EudraVigilance (European Union Drug Regulating Authorities Pharmacovigilance) came into being as an official European database for managing and analyzing suspected adverse reactions from clinical trials.
2004: Canadian health officials implemented Good Pharmacovigilance Practices (GVP).
2007: Passed the Food and Drug Administration Amendments Act.
The Prescription Drug User Fee Act (PDUFA) and the Medical Device User Fee and Modernization Act (MDUFMA) reauthorized and expanded.
In September 2008, the European Commission published Volume 9A pharmacovigilance guidance for human medicinal products.
In September 2010, the FDA issued final regulations addressing the safety reporting requirements for investigational new drug applications (INDs).
December 2010 – New PV legislation adopted – Updated the EudraVigilance Medicinal Product Dictionary (EVMPD).
11 June 2011 – the Commission published guidance on
- collection, verification, and presentation of adverse reaction reports arising from clinical trials on medicinal products for human use (CT-3)
July 2012 – EMA passed new pharmacovigilance legislation and replaced Volume 9A with good pharmacovigilance practice guidelines.
2014: EMA and FDA formed a cluster relationship to participate in collaborative processes for information-sharing on GVP. This relationship aims to share data, research, and coordinate other activities.
November 2017 – Formulation of new EudraVigilance.
January 2018 – India published the Pharmacovigilance Guideline Document for Marketing Authorization Holders of Pharmaceutical products.
One thought on “Historical Evaluation of Pharmacovigilance”